
华南海鲜市场不是病毒发源地!中科院团队基因追踪“零号病人”再进一步
时间 : 2020-02-22 15:38:59
来源 : 科普时报社
【字体:大 中 小】
【打印】
华南海鲜市场的新冠病毒来自其他地方,这个结论有了基于基因序列的证据。
中国科学院西双版纳热带植物园的研究人员在分析 93 个新型冠状病毒样本基因组数据后发现,基于 120 个变异位点得到 58 种单倍型(基因类型)中,来自华南海鲜市场患者样品单倍型与 H1 有关,而作为更古老的基因类型样本 H3、H13 和 H38 则来自华南海鲜市场之外。这印证了华南海鲜市场不是病毒发源地的推论。
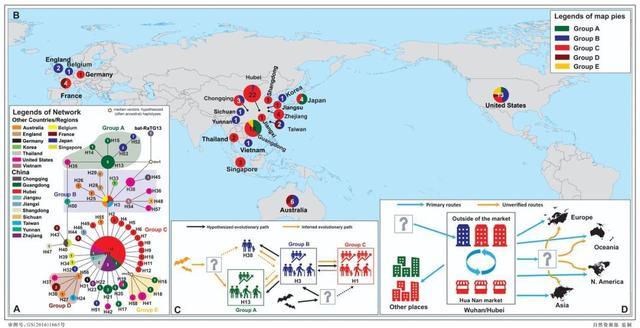
(图 | 新型冠状病毒 58 种单倍型的演化关系和地理分布格局(A,B),单倍型之间的可能演化关系(C),以及新型冠状病毒的可能传播和扩散路线(D);A 和 B 圆圈中的数据是样本数量(来源:郁文彬)
这个研究印证了此前 1 月 24 日发表在《柳叶刀》的研究结论,即追踪第一批 41 个确诊病例发现,这批最早在 12 月 1 日报告的病例中,总共有 13 个病例与海鲜市场无接触史。也就是说,病毒在进入海鲜市场之前就已经潜入武汉居民的生活中。
这个研究由中国科学院西双版纳热带植物园郁文彬联合华南农业大学和北京脑科中心的科研人员进行,于 2 月 19 日发表在中国科学院科技论文预发布平台 ChinaXiv 上。
作者认为,确定海鲜市场是不是病毒发源地对于寻找病毒的来源,以及确定中间宿主,对疫情的控制和避免再次爆发具有至关重要的意义。
全基因组数据分析溯源
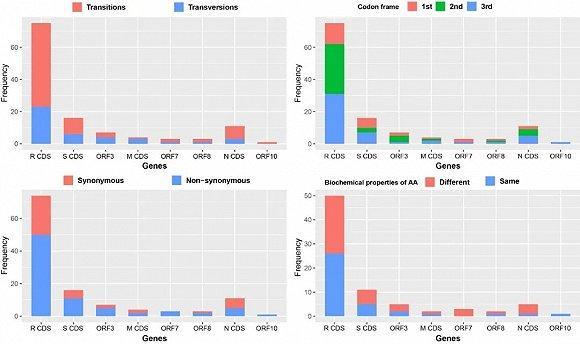
图 | 跨越 8 个编码区的 120 核苷酸突变分析(来源:郁文彬)
郁文彬对 DeepTech 解释说,这是一种基于种群遗传学的分析方法。一种基因型不等于一个患者,一种新冠病毒单倍型携带者有可能是多人,甚至可能是上百人,比如 H1 单倍型就对应了 19 个样本,但是可以通过这个来溯源,因为某个单倍型患者意味着可能是来自同一个源传染。
研究人员收集了全世界各领域共享到 GISAID EpiFluTM 数据库中的 93 个新型冠状病毒样本的基因组数据(截止 2 月 12 日),覆盖了四大洲 12 个国家。其中有 39 个样本来自 11 个国家的感染患者,编码了 31 个单倍型。来自中国的 54 个样本也编码了 31 个单倍型。
他们通过全基因组数据解析,追溯传染源及扩散路径发现,收到的 93 个样本包含 58 种单倍型,而 58 个单倍型中,H13 和 H38 是比较“古老的”单倍型,两者通过单倍型 H3 衍生出了单倍型 H1。
值得注意的是,与华南海鲜市场有关联的患者样品单倍型都是 H1 及其衍生单倍型,而一份来自武汉样品的更“古老的”单倍型 H3 则与华南海鲜市场无关。作者结合第一批患者报告信息,以及种群扩张时间分析结果,作者推测华南海鲜市场的新型冠状病毒是从其他地方传入进来,在市场中发生快速传播蔓延到市场之外。
作者还推测,单倍型 H13 和 H38 通过一个中间载体与蝙蝠冠状病毒 RaTG13 关联,这个中间载体 mv1 可能是一个祖先单倍型,也可能来自中间宿主或者“零号病人”。
“古老的”单倍型 H13 和 H38 的病毒样品分别来自深圳的病患(广东首例)和美国华盛顿州的病患(美国首例)。他们的旅行记录表明 2019 年 12 月底至 2020 年 1 月初都来过武汉。
为什么有 2 个“古老的”单倍型呢?郁文彬对 DeepTech 解释说,其实 H13 和 H38,以及 H3,它们两两之间只有 1 个核苷酸位点的差异,有可能是衍生到 H3 后,发生了一次转换的回复突变,衍生到另外一种基因型基因型。并且两者都是同义突变,即该突变不影响基因表达的结果。这就意味着 2 个“古老的”单倍型极为接近。
现有武汉样本中没有检测到 H13 和 H38 单倍型,作者推测可能是因为现有样品主要采自几家定点医院,而且样品采集时间局限于 2019 年 12 月 24 日和 2020 年 1 月 5 日。那么,如果能在武汉其他医院早期的病患中检测到这两种单倍型,将对于寻找病毒来源非常有帮助。
对于单倍型 H3,其武汉样本的发现时间在 2020 年 1 月,但这并不意味着其基因型更“年轻”。郁文彬解释说,可能是因为病毒在被传染到这位携带者身上没有发生变异,或者很早传染到这位患者,只是没有发病。
追踪“零号病人”
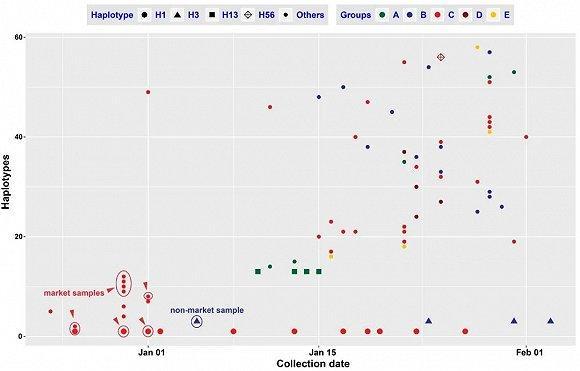
图 | 新型冠状病毒单倍型的样品采集时间情况。红色圈的样品是确认与华南海鲜市场有关;蓝色圈的样本确认与海鲜市场无关(来源:郁文彬)
华中农业大学一位匿名教授评价说,这是一项基于现有新型冠状病毒基因组序列的研究。理论上根据其变异的情况,是能够溯源各个样本之间的关系,到底是父子关系还是兄弟姐妹关系,到底复制了多少代。那么就可以最终追溯出最早的新冠病毒从哪里来。当然这里有一个假设是病毒是从动物传播到人。
在华南海鲜市场之外发现单倍型 H3 也就意味着,H3 携带者传染了去华南海鲜市场的人,结果将人流拥挤的华南海鲜市场变成了病毒传播爆发点。
他认为,溯源很重要,这个研究是通过大数据分析的,非常有意义。最核心的调查是最早感染新冠病毒的那几个人,这尤其需要医院配合,但是医院可能现在没心思、没精力来做这件事,那些医生可能也没有进化基因组学的背景,所以他们也可能没有认识到这件事的价值。
他说,至于这个最原始的携带者,也就是“零号病人”,可能是接触蝙蝠的人。理论上还是能够追溯的,即使零号病人已经去世,在某个医院应该还保留了他的血液样本。
郁文彬认为,最原始的单倍型不太可能存在现在的患者身上,因为病毒的变异太快。他们现在能拿到的数据已经是最早是 12 月 24 日,因为没有早期的样本就很难去溯源,“我们猜测国内医院可能没有保存早期患者的组织材料,当然我们非常希望有早期的样品存在”。
郁文彬表示,这个研究有个不足,那就是 93 个样本中,武汉样本主要是早期的,如果有更多武汉样本进行基因组测序的话,可能在溯源方面可以找到更多的证据,比如说找到 H13 和 H38 单倍型,可能可以帮助找到病毒来源。
一个好消息
郁文彬说,这项研究主旨是分析整个新冠病毒基因组的变异情况,以及弄清楚新冠病毒传播式样,基于传播和扩散的分析,为寻找病毒来源提供基因上的证据。
研究人员发现新型冠状病毒基因组没有发生重组事件,93 个基因组之间有 120 核苷酸发生了突变(0.41% 序列长度),并均匀分散在 10 个编码区。120 个突变的核苷酸关联了 119 个氨基酸密码子,其中 79 个密码子(65.83%)改变了氨基酸类型,并有 42 个(53.17%)氨基酸理化性质都被改变。
“新冠病毒基因组尚未发生重组事件”的结论是一个好消息,也就是说不会产生新的全新的病毒毒株。
研究的另一个结论是,新冠病毒在 2 月 12 日之前发生过 2 次明显的种群扩张。
根据新型冠状病毒基因组发生重组时间推算,1 月之前的种群扩张发生时间是 12 月 8 日,那么病毒可能在 12 月初,甚至 11 月下旬即已经开始有人际传播,随后在华南海鲜市场加快了人际传播。
研究推算 2 月份之前的种群扩张时间在 1 月 6 日,这个可能与元旦假期有关联。需要指出,此时国家疾控中心发布了 2 级应急响应。
研究团队认为,当时的预警起到了一些警示作用,公众活动和出行都有所减少。如果当时的警示能引起大众更广泛的重视,那么 1 月份中下旬向全国和全球蔓延的病例会有所降低。